Craniodiaphyseal Dysplasia
What is Craniodiaphyseal Dysplasia?
Craniodiaphyseal dysplasia abbreviated as CDD is also called lionitis, which refers to a very rare autosomal genetic bone disorder leading to the deposition of calcium going to the skull, facial features disfigured and it can reduce the life expectancy of a person.
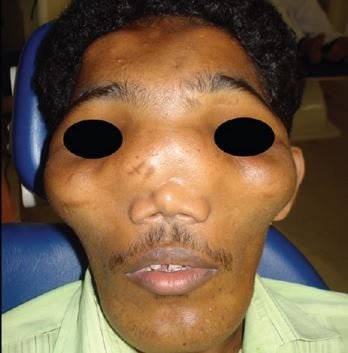
A lot of calcium deposit affects the cranial foramina by decreasing its size and at the same time, it has the capacity to decrease the size of the hole located in the cervical spinal canal. Patients having this bone problem usually die during childhood.
This bone disease is characterized by an excessive bone growth and hardening of some tissues, which specifically affects the skull bones and facial bones. Studies have recorded of only 30 cases of existing CDDs.
Signs and Symptoms
The calcium build-up in a person’s skull reduces both the size of the cranial foramina and the cervical hole of the spinal canal. Because of these changes, cranial nerves are compressed, to include foramen magnum as well as the intracranial contents.
The compression of the cranial nerves and the thickening of skull bones can lead to some signs and symptoms which are:
- Facial paralysis
- Blindness
- Difference in the size of the eyes
- Hearing impairment
- Abnormality of the ribs
- Epilepsy
- Mental retardation
- Increased intracranial pressure
- Short stature
- Macrocephaly
- Cognitive impairment
- Sclerosis of the expanded diaphyses
- Hyperostosis in long bones
- Postural defect in the metaphyses
Some of the complications of CDD are severe facial deformity, facial diplegia, complete hearing inability, nasolacrimal obstruction, quadriparesis, significant brain compression, mechanical damage of nerve fibers and deformation of ossicles due to bony overgrowth.
Causes of Craniodiaphyseal Dysplasia
CDD is a hereditary disorder, where the underlying causative gene remains to be unknown yet. The problem is seen to be acquired through an autosomal recessive trait. There are only a very few cases of CDD which have been reported, which links to the difficulty in proving its genetic association.
Diagnosis
Diagnosis of patients with CDD is performed by physicians through assessing for the presence of some clinical symptoms, or through a CDD differential diagnosis.
The very common reported sign for an existing CDD is the difference in the size of both eyes of a person. Other clinical manifestations to be checked are the presence of paralysis in the body, moments of seizures, characteristics of a mentally retarded person and an infection of the lacrimal sac.
Differential diagnosis to confirm and eliminate CDD apart from other disease conditions is through the mutation analysis of the TGFB1 gene, where physicians can rule out a similar problem with CDD which is the Camurati-Engelmann disease. Other problems taken into consideration are Van Buchem’s dysplasia and craniotubular dysplasias.
Prenatal diagnostic testing is not available maybe due to the very low incidence of the condition among patients.
An X-ray scan may show possible areas with an unusual bone density, an excessive deposition of calcium in affected skeletal structures and growing numbers of osteoblasts, and cells which are developing new bones might also be observed.
Treatment
Patients who suffer with CDD might be recommended to undergo surgery to relieve symptoms of decompression; however, there can be chances that the craniodiaphyseal dysplasia may replace the removed bone after the surgical procedure, and the same problem can recur anytime.
It must always be remembered that this surgical option has significant risks due to the fact that delicate structures are being involved.
CDD management can be done through:
- Patients have to be taken to an anesthetist when they encounter problems with tracheal intubation and with airway management.
- Decompressing craniectomy can be done to aid in an acute brain decompression, with the purpose of enlarging the anterior and middle fossa in cases where there are signs of an elevated intracranial pressure.
- Decompression of the optic nerve and orbital nerve might be needed to resolve cases of papilledema among patients.
- Calcitriol/calcitonin therapy may be tried to reduce the clinical course of CDDs.
- Prednisone treatment is used with Calcitriol/calcitonin therapy.
- Calcium diet can also be used with Calcitriol/calcitonin therapy.
The long-term treatment is performed by attending physicians with the aim to control the rapid malformation of skeletal bones. Patients with CDDs are closely monitored to manage the progressive signs of the bone problem.
Pictures
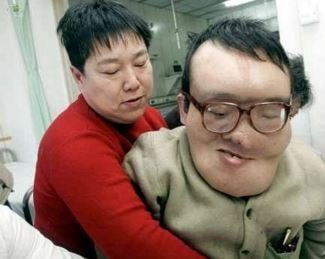
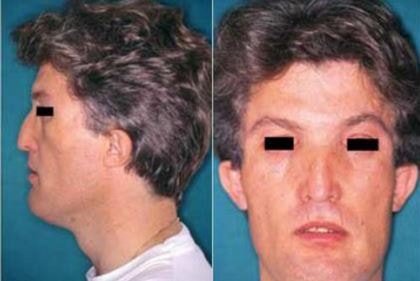

Prognosis
The prognosis of CDD is poor despite some management done. The advantages of performing craniofacial remodeling, choanal stenosis surgery and dacryocystorhinostomy among patients is only good for a short period. Papilledema might recur and there is the possibility of a rapid reconstitution of the osseous optic canals of the affected eyes.
Patients live shortly and usually die during childhood years so that life expectancy is considered bad for them in terms of chances for survival. It is always best to seek medical attention at an early phase of life, particularly even during infancy period, so that earlier management can be started to control and prevent the progression of the problem into its worst case.
References
- http://www.primehealthchannel.com/craniodiaphyseal-dysplasia.html
- http://www.rightdiagnosis.com/c/craniodiaphyseal_dysplasia/intro.htm
- https://rarediseases.info.nih.gov/gard/1567/craniodiaphyseal-dysplasia/resources/9
- Brueton LA, Winter RM (November 1990). “Craniodiaphyseal dysplasia”. J. Med. Genet. 27 (11): 701–6. doi:10.1136/jmg.27.11.701. PMC 1017262. PMID 2277386.
- McKeating JB, Kershaw CR. Craniodiaphyseal dysplasia. Partial suppression of osteoblastic activity in the severe progressive form with calcitonin therapy. J R Nav Med Serv. 1987 Summer; 73(2):81–93.
- Schaefer B, Stein S, Oshman D, Rennert O, Thurnau G, Wall J, Bodensteiner J, Brown O. Dominantly inherited craniodiaphyseal dysplasia: a new craniotubular dysplasia. Clin Genet. 1986 Nov; 30(5):381–391.